|
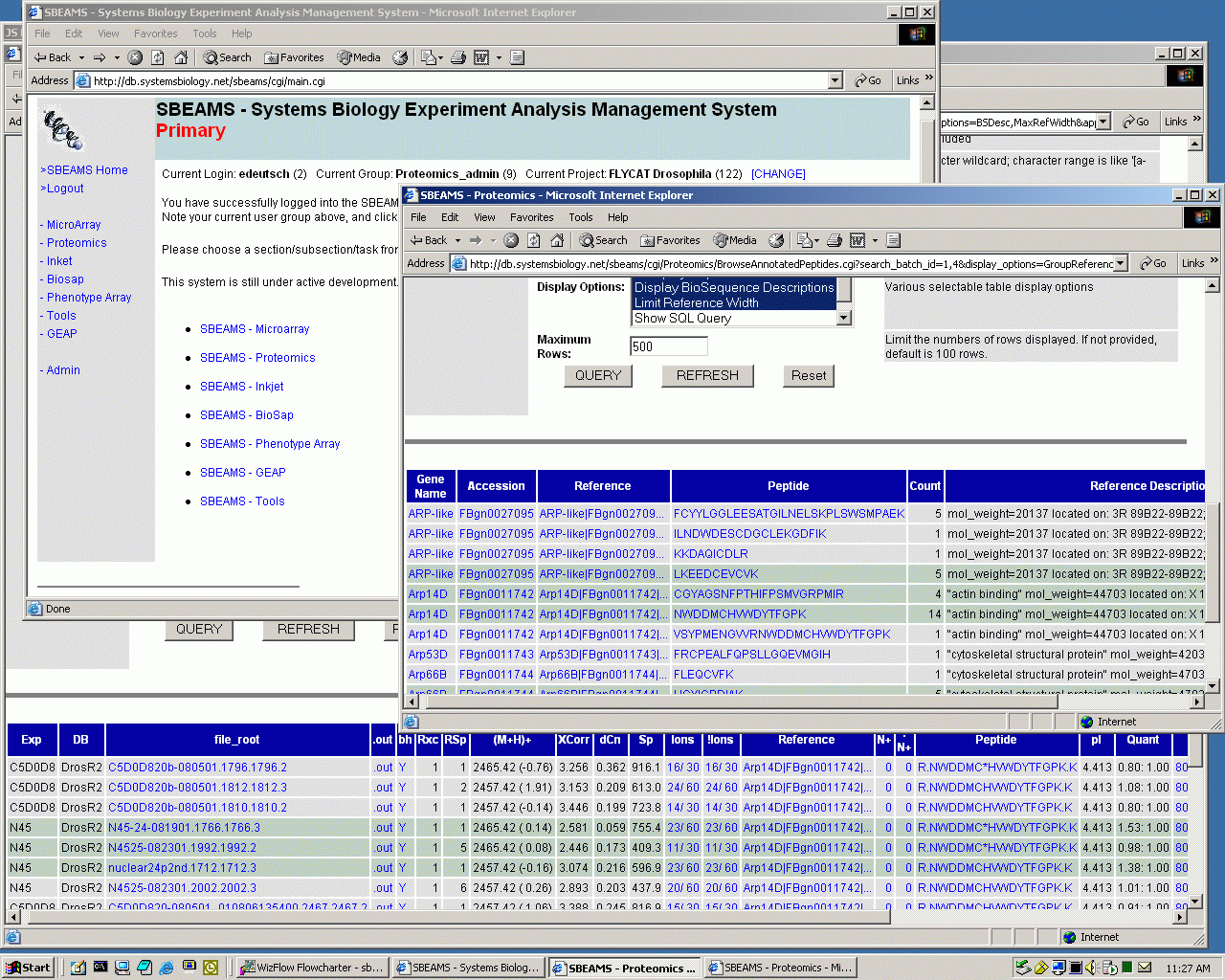
SBEAMS - Proteomics OverviewThe current SEQUEST analysis sequence of Proteomics data is completely file based. Raw data files are generated by the instrument. Individal mass spectra are extracted from the data files and are searched with Sequest which in turn creates more output files. These files are summarized in two steps to create a single HTML result. This final file is iteratively culled by the investigator to filter out peptides which are either misidentified or perhaps uninteresting. This scales well with respect to disk space and doesn't require much CPU power (except for the Sequest step, of course) since it is a simple process. A filesystem imposes little order and can thus be both very flexible and very messy. However, it becomes very difficult to keep track of many experiments this way, integrate results from multiple experiments, and capitalize on the analysis performed in previous experiments while analyzing a new one. To address these needs, we are developing software which uses a relational database management system (RDBMS) to manage the organization, storage, and exploration of the Proteomics data produced at ISB. SBEAMS-Proteomics is part of the SBEAMS (Systems Biology Experiment Analysis Management System) Project, which is a framework for collecting, storing, and accessing data produced by a variety of different experiments; these experiments can be managed separately but then correlated later under the same framework. This integrated system is a consistent framework that combines a unified state-of-the-art relational database management system (RDBMS) back end, a collection of tools to store, manage, and query experiment information and results in the RDBMS, a web front end for querying the database and providing integrated access to remote data sources, and an interface to existing programs for clustering and other analysis. Since all data from each step of the experiment are warehoused in a modular schema in the RDBMS, quality control and data analysis tasks are greatly simplified. With SBEAMS-Proteomics, users can first enter their project, experiment, and sample information into the database. The data products of the file-based analysis pipeline are then ingested into the database, and subsequent exploration of the data, annotation of individual pieces, and correlation with other experiments can all be managed within the same system. The interface allows a much more flexible analysis of the data: individual genes, proteins, and peptides may be examined closely across multiple experiments; information about the quality of identification can be stored with the data; peptides which could not be properly identified from high quality spectra and be flagged and followed up with additional searches. The SBEAMS framework makes it easy to add a quick interface to new queries to explore the data. The knowledge stored while analyzing experiments remains easily accessible at later times and by other investigators to fully capitalize on previous work. There are disadvantages associated with such a system, and nearly all are associated with running the RDBMS. An RDBMS capable of easily ingesting and providing quick query results on many experiments of several hundred megabytes each requires significant hardware. A machine with 4-8 CPUs, 4-8 GB of RAM, and 500-1000 GB of database storage is really needed to keep up with several experiments produced per week and multiple users accessing the data. For just a few users and experiments, today's commoditity dual CPU hardware would suffice. Managing a large database requires more administrative talent and time than managing a large filesystem. And finally software that interacts with a RDBMS adds a layer of complexity over programs which just manipulate flat text files. Noneless, these disadvantages are certainly outweighed by the new capabilities afforded by SBEAMS-Proteomics. Screenshots and diagrams:
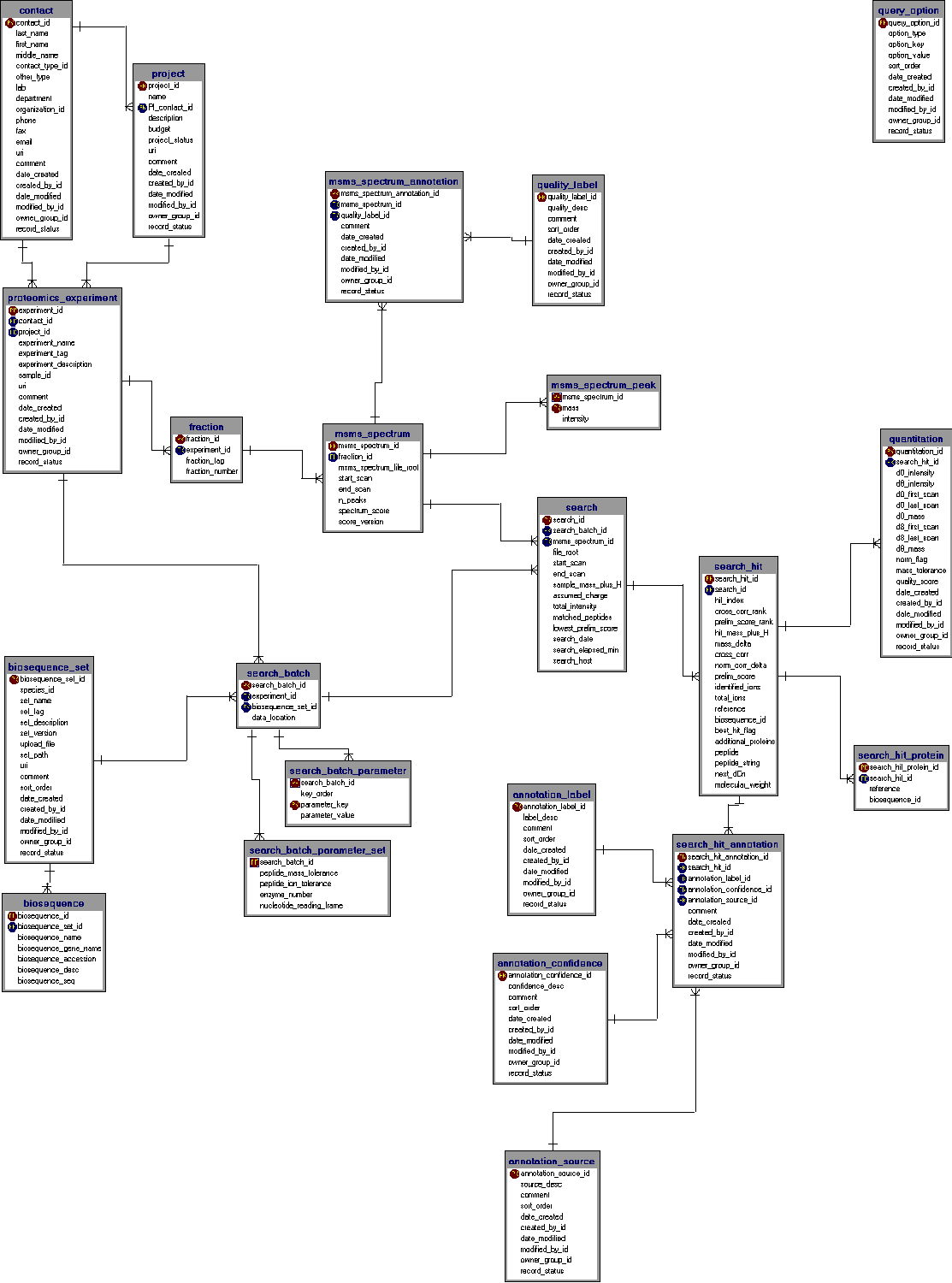
Below that to the right is a window in which the user has selected to issue a SQL query (query parameter entry fields are scrolled out of view) that summarizes the peptides that have been annotated for the "ARP*" genes in two Drosophila Proteomics experiments (click here to execute this query on the database - SBEAMS login required). Many peptides have been observed and annotated just once, while several have been annotated many times. Various hyperlinks give the user access to more information about the genes, proteins, and peptides actually observed and annotated. At the bottom is additional information about all the annotated occurrences of one of the peptides as identified by SEQUEST. The table includes information about which of the two experiments the peptides were observed in, the masses, the actual peptides (some of which contain tagged cysteines), pI values, ICAT quantitation ratios, annotation information (clipped off right edge) and much more. Search results can be annotated to additional insights. Here is the current in-development relational schema for the
SBEAMS-Proteomics database:
| |||||||